Independent Principal Component Analysis (IPCA)
In some case studies, we have identified some limitations when using PCA:
- PCA assumes that gene expression follows a multivariate normal distribution and recent studies have demonstrated that microarray gene expression measurements follow instead a super-Gaussian distribution
- PCA decomposes the data based on the maximization of its variance. In some cases, the biological question may not be related to the highest variance in the data
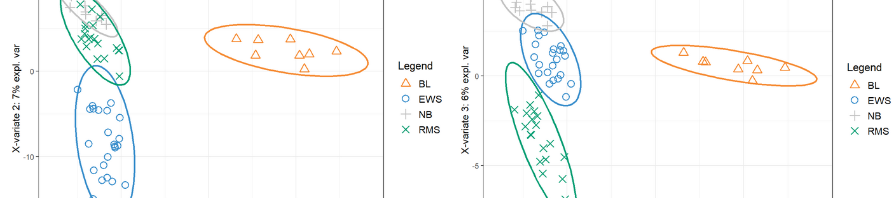
Instead, we propose to apply Independent Principal Component Analysis (IPCA) which combines the advantages of both PCA and Independent Component Analysis (ICA). It uses ICA as a denoising process of the loading vectors produced by PCA to better highlight the important biological entities and reveal insightful patterns in the data.
IPCA offers a better visualization of the data than ICA and with a smaller number of components than PCA.
How to choose the number of components:
The kurtosis measure is used to order the loading vectors to order the Independent Principal Components. We have shown that the kurtosis value is a good post hoc indicator of the number of components to choose, as a sudden drop in the values corresponds to irrelevant dimensions.
Sparse Independent Principal Component Analysis (sIPCA)
Similar to the [intlink id=”129″ type=”page”]sparse PCA[/intlink] version implemented in mixOmics, soft-thresholding is applied in the independent loading vectors in IPCA to perform internal variable selection.
How to choose the number of variables to select:
The number of variables to select is still an open issue. In our paper we proposed to use the Davies Bouldinmeasure which is an index of crisp cluster validity. This index compares the within-cluster scatter with the between-cluster separation.
More details about how to use the ipca.R function in the[intlink id=”233″ type=”page”] case study[/intlink].
References